Let’s say you have run the DNA of your study species on a next generation sequencing machine, such as Illumina HiSeq. You now have a huge collection of short sequences to make sense of. The data is a text file that is too big to open in a regular text editor.
To have a look at the raw data, type:
less [name_of_data_file]
The format (fastq) tends to evolve, but lately it looks something like this:
@HWI-962:55:C0A4UACXX:4:1101:1375:2341 1:N:0:TCTCAG
GTCACTGATCGGGGAATTCGTACAGTCTCGTATATGTGAGTCAATGCGTCGTNGCTCCCCTTAACG
TACGTGCAATCTTGTCGGTTCGGCGGCACAAAA
+
B?BDFDDFHHHHHJJJDHHIEFGGGCGBFF6DFH@HBFHI9B@FDHIJBHH;#-5;CHFF;@@CED?BBD(5AACDACDDCCDD59@@BBBDB-9ABDB
The first bit includes the machine name and information of the run and position of the cluster on the flowcell. The first line ends with the barcode sequence. Then follow nucleotide sequence and a quality score. The quality scores can be in various formats.
You will want to have a closer look at the data quality before you proceed. The fastcq toolkit is very useful for this.
It generates quality plots like this:
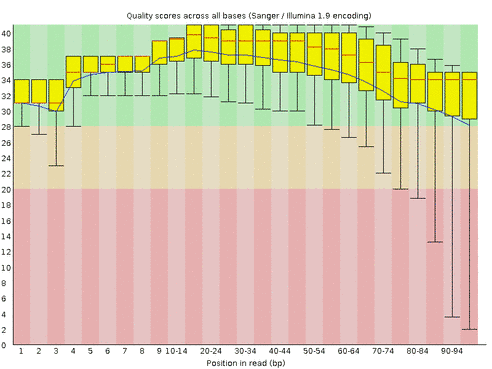
The quality of the first few bases is slightly lower, because the machine starts sequencing at lower intensity, which allows it to locate the clusters more unambiguously. You’ll also notice that the quality drops off at the end of the read, which is due to reagents getting old. This is normal, and in this case it stays within an acceptable range. However, some trimming or clipping is often necessary. The fastx toolkit can be used for this. Alternatively, you can use a package that does all the trimming, clipping, adapter removal etc automatically. Many of these tend to be quite harsh. I tend to use Trimmomatic, which seems to do a good job. The command line looks a bit messy. Here is an example:
java -classpath /usr/local/Trimmomatic/Trimmomatic-0.17/trimmomatic-0.17.jar org.usadellab.trimmomatic.TrimmomaticPE -phred33 read1.fastq read2.fastq read1_forward_paired.fq read1_forward_unpaired.fq read2_reverse_paired.fq read2_reverse_unpaired.fq ILLUMINACLIP:illuminaClipping.fa:2:40:15 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:36